November 2019 Newsletter: Tubulin Hyperglutamylation, Mitochondria, and Neurodegeneration
Microtubules (MTs) are composed of α/β-tubulin heterodimers and are one of three essential proteins that comprise the cytoskeleton of mammalian cells and have essential roles in cell development, growth, motility, mechanotransduction, and intracellular trafficking. Functional regulation of MTs is achieved through at least seven different post-translational modifications (PTMs) that usually occur post-polymerization and preferentially on the α/β-tubulin heterodimers of stable (vs dynamic) MTs. PTMs are highly dynamic and often reversible processes that regulate a protein’s functions, binding partners, and/or subcellular localization by addition of a chemical group or a peptide to amino acid residue(s) within the target protein1-4.
The polyglutamylation PTM, addition of variable length glutamate side-chains to primary sequence glutamate residues, was first described in the early 1990s. Glutamate residues in the C-terminal tails (CTTs) of α- and β-tubulins are the most common substrates1-6 (Fig. 1). Glutamylation enzymes are members of the tubulin tyrosine ligase-like (TTLL) family of proteins1-4,7,8. Cytosolic carboxypeptidases (CCP; a.k.a. Nna) function as deglutamylases with CCP1, 4, 5, and 6 removing glutamate side-chain residues in mammalian cells. Three enzymes (CCP1, CCP4, and CCP6) catalyze the shortening of polyglutamate chains, while CCP5 specifically removes the branching point glutamate residue1-4,9-11 (Fig. 1). Physiological polyglutamylation modifies MTs within neuronal cell bodies and processes (i.e., dendrites and axons) to regulate a variety of MT-based neuronal functions1-4.
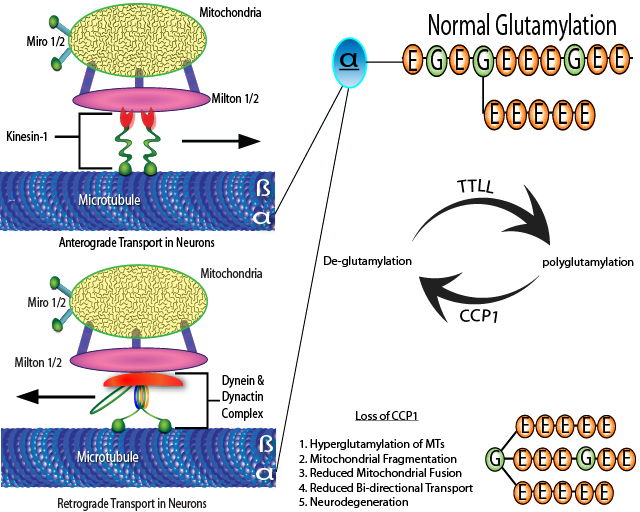
Figure 1. Schematic representation of the bi-directional transport of mitochondria in neurons and effects of hyperglutamylation due to loss of CCP1 activity. The anterograde motor kinesin-1 and the retrograde motor dynein/dynactin complex directly interact with Miltons and Miros on mitochondria to drive their transport along MTs. Functional loss of CCP1, a deglutamylation enzyme, results in hyperglutamylation which negatively affects mitochondrial dynamics and results in neurodegeneration. The carboxyl-terminal amino acids of tubulin can be acted upon by TTLLs (glutamylation enzymes) and CCPs (deglutamylation enzymes), including CCP1. CCP1 cleaves glutamate residues from the polyE side chain down to the branch point. Inactivating mutations in CCP1 are responsible for Purkinje neuron degeneration in pcd mice. This figure is adapted from those in references 9 and 17.
Neurodegeneration and Mitochondria
Hyperglutamylation of tubulin can cause neurodegeneration, first observed in the death of Purkinje cells in pcd (Purkinje cell degeneration) mice due to inactivating mutations in CCP19,12. Purkinje neuron death was prevented and motor coordination improved following down-regulation of the tubulin-specific neuronal polyglutamylase (TTLL1) in the cerebellum of young pcd mice9. A recent study using Purkinje-cell-specific Ccp1-knockout mice confirmed not only that tubulin hyperglutamylation due to CCP1 inactivation can cause neurodegeneration, but that it does so by disrupting MT-based intraneuronal transport13,14. Mice lacking both Ccp1 and ttll1 did not display Purkinje cell death13,14. Even more intriguing is a mechanistic study which revealed how loss of CCP1 expression/activity causes neurodegeneration in pcd mice14,15 (Fig. 1). Using primary cerebellar neurons cultured from pcd mice and CCP1-deficient retinal pigment epithelial cells (involved in retinal degeneration of pcd mice) produced the expected increase in tubulin polyglutamylation14,15. Furthermore, loss of Ccp1 reduced mitochondrial fusion and bi-directional (i.e., anterograde and retrograde) motility, as well as increased instances of mitochondrial fragmentation14,15. As with any preclinical models of neurodegeneration, the observation of genetic mutations in human subjects cements the importance of studying the disease not just from the perspective of learning more about neurodegenerative diseases, but to also devise therapeutic interventions. Recently, Shashi et al.16 demonstrated that loss of CCP1 enzymatic activity (via either loss-of-function variants or missense variants with single amino acid changes) is responsible for a previously unexplained childhood-onset, progressive neurodegenerative condition.
Taken together, the above studies indicate that intraneuronal transport of mitochondria and maintenance of mitochondria functionality and morphology are compromised by tubulin hyperglutamylation. Importantly, mitochondria provide the energy needed for intracellular bi-directional transport and maintenance of ion gradients in neurons, two processes required for basic neuronal functions, including synaptic neurotransmission, synaptic vesicle recycling, and intracellular trafficking. Mitochondria are highly dynamic organelles that undergo continuous fusion, fission, and transport, processes which not only control mitochondrial morphology and number but also regulate mitochondrial function and location. Mitochondria themselves rely upon fast bi-directional intracellular transport along MTs to localize to specific cellular locations17 (Fig. 1). The primary components of the motor-adaptor complex are the anterograde motor kinesin-1 (a.k.a. kinesin heavy chain [KHC] or KIF5), retrograde motor dynein (in complex with dynactin), Miro1 and 2, and Milton1 and 217. Milton isoforms are mitochondrial adaptor proteins that recruit the molecular motors to mitochondria for MT-mediated transport and Miro isoforms are mitochondrial Rho GTPases. The anterograde motor kinesin-1 and the retrograde motor dynein/dynactin complex directly interact with Miltons and Miros on mitochondria to drive their bi-directional movement along the MTs18,19.
An important question is if these findings extend to other neurodegenerative diseases? It is already well known that pathogenic tubulin PTMs, including those that affect binding of MAPs (microtubule-associated proteins), play a key role in Parkinson’s disease (PD) and various tauopathies12,20. Roles for MT polyglutamylation in other neurodegenerative diseases remains an open question, but mitochondrial dysfunction is observed in PD, Alzheimer’s disease (AD), Huntington’s disease (HD), and amyotrophic lateral sclerosis (ALS)17. The importance of understanding the roles of mitochondrial dynamics in the pathogenesis of neurodegenerative diseases has intensified after the identification of several key regulators (e.g., Drp1, OPA1, mitofusins) of mitochondrial fusion and fission. Furthermore, there is a clear need to examine the possible interplay between tubulin PTMs, mitochondrial dysfunctions, and neurodegenerative diseases17.
Summary
Despite recent gains in understanding tubulin polymodifications, much remains to be discovered, including identification of non-enzymatic regulators of glutamylation/deglutamylation (e.g., CSAP [cilia and spindle-associated protein])21 and all tubulin deglutamylases, as well as a complete understanding of how glutamylation (and other PTMs) affect binding of MAPs (e.g., tau, molecular motors)9-11,21 and the activity of MT-severing enzymes21. Identification of the complete population of glutamylation and deglutamylation enzymes and related regulators will reveal new therapeutic targets for neurodegenerative diseases that have dysfunctional intracellular transport as a pathophysiological hallmark. To assist researchers, Cytoskeleton offers a variety of Signal-Seeker Detection Kits which allow the measurement of endogenous levels of PTMs, tubulin activity and binding assay kits, live cell imaging probes for microtubules, and purified tubulin proteins.
References
- Hammond J. et al. 2008. Tubulin modifications and their cellular functions. Curr. Opin. Cell Biol. 20, 71-76.
- Janke C. and Kneussel M. 2010. Tubulin post-translational modifications: encoding functions on the neuronal microtubule cytoskeleton. Trends Neurosci. 33, 362-372.
- Wloga D. and Gaertig J. 2010. Post-translational modifications of microtubules. J. Cell Sci. 123, 3447-3455.
- Wloga D. et al. 2017. Tubulin post-translational modifications and microtubule dynamics. Int. J. Mol. Sci.18, 2207.
- Edde B. et al. 1990. Posttranslational glutamylation of alpha-tubulin. Science. 247, 83-85.
- Nogales E. 2000. Structural insights into microtubule function.Annu. Rev. Biochem. 69, 277-302.
- Janke C. et al. 2005. Tubulin polyglutamylase enzymes are members of the TTL domain protein family. Science. 308, 1758-1762.
- van Dijk J. et al. 2007. A targeted multienzyme mechanism for selective microtubule polyglutamylation.Mol. Cell. 26, 437-448.
- Rogowski K. et al. 2010. A family of protein-deglutamylating enzymes associated with neurodegeneration. Cell. 143, 564-578.
- Kimura Y. et al. 2010. Identification of tubulin deglutamylase among Caenorhabditis elegans and mammalian cytosolic carboxypeptidases (CCPs). J. Biol. Chem. 285, 22936-22941.
- O’Hagan R. et al. 2011. The tubulin deglutamylase CCPP-1 regulates the function and stability of sensory cilia in C. elegans. Curr. Biol. 21, 1685-1694.
- Baird F.J. and Bennett C.L. 2013. Microtubule defects and neurodegeneration. J. Genet. Syndr. Gene Ther. 4, 11.
- Magiera M.M. et al. 2018. Excessive tubulin polyglutamylation causes neurodegeneration and perturbs neuronal transport. EMBO J. 37, e100440.
- Strzyz P. 2018. Neurodegenerative polyglutamylation. Nat. Rev. Mol. Cell Biol. 20, 1.
- Gilmore-Hall S. et al. 2018. CCP1 promotes mitochondrial fusion and motility to prevent Purkinje cell neuron loss in pcdmice. J. Cell Biol. 218, 206-219.
- Shashi V. et al. 2018. Loss of tubulin deglutamylase CCP1 causes infantile-onset neurodegeneration. EMBO J. 37, e100540.
- Gao J. et al. 2017. Abnormalities of mitochondrial dynamics in neurodegenerative diseases. Antioxidants. 6, 25.
- Guo X. et al. 2005. The GTPase dmiro is required for axonal transport of mitochondria to drosophila synapses. Neuron. 47, 379–393.
- Glater E.E. et al. 2006. Axonal transport of mitochondria requires milton to recruit kinesin heavy chain and is light chain independent. J. Cell Biol.173, 545–557.
- Pellegrini L. et al. 2017. Back to the tubule: microtubule dynamics in Parkinson’s disease. Cell. Mol. Life Sci. 74, 409-434.
- van der Laan S. et al. 2019. Tubulin glutamylation: a skeleton key for neurodegenerative diseases. Neural Regen. Res.14, 1899-1900.
Related Products
Fluorescent Tubulins
Tubulin protein (fluorescent AMCA): porcine brain (Cat. TL440M)
Tubulin protein (fluorescent HiLyte 488): porcine brain (Cat. # TL488M)
Tubulin protein (rhodamine): porcine brain (Cat. # TL590M)
Tubulin protein (X-rhodamine): bovine brain (Cat. # TL620M)
Tubulin protein (fluorescent HiLyte 647): porcine brain (Cat. # TL670M)
Signal Seeker™ Kits
Signal-Seeker™ Phosphotyrosine Detection Kit (30 assay) (Cat. # BK160)
Signal-Seeker™ Phosphotyrosine Detection Kit (10 assay) (Cat. # BK160-S)
Signal-Seeker™ Ubiquitination Detection Kit (30 assay) (Cat. # BK161)
Signal-Seeker™ Ubiquitination Detection Kit (10 assay) (Cat. # BK161-S)
Signal-Seeker™ SUMOylation 2/3 Detection Kit (30 assay) (Cat. # BK162)
Signal-Seeker™ SUMOylation 2/3 Detection Kit (10 assay) (Cat. # BK162-S)
Signal-Seeker™ Acetyl-Lysine Detection Kit (30 assay) (Cat. # BK163)
Signal-Seeker™ Acetyl-Lysine Detection Kit (10 assay) (Cat. # BK163-S)
NEW Signal-Seeker™ SUMOylation 1 Detection Kit (30 assay) (Cat. # BK165)
NEW Signal-Seeker™ SUMOylation 1 Detection Kit (10 assay) (Cat. # BK165-S)
Tubulin Kits
Tubulin polymerization HTS assay using >97% pure tubulin, OD based - Porcine (Cat. # BK004P)
Tubulin polymerization assay using >99% pure tubulin, OD based - Porcine (Cat. # BK006P)
Tubulin polymerization assay using >99% pure tubulin, fluorescence based (Cat. # BK011P)
Microtubule Binding Protein Spin-Down Assay Biochem Kit (Cat. # BK029)
Microtubule/Tubulin In Vivo Assay Biochem Kit (Cat. # BK038)
Live Cell Imaging Products
